|
|||||||
 |
|||||||

|
||
This document is published here with the kind permission of the author, Dr.Prof. Massimo Castagnaro DVM, PhD, ECVP of Turin University, and the editor of "Acta Neuropathologica", where this document was first printed.
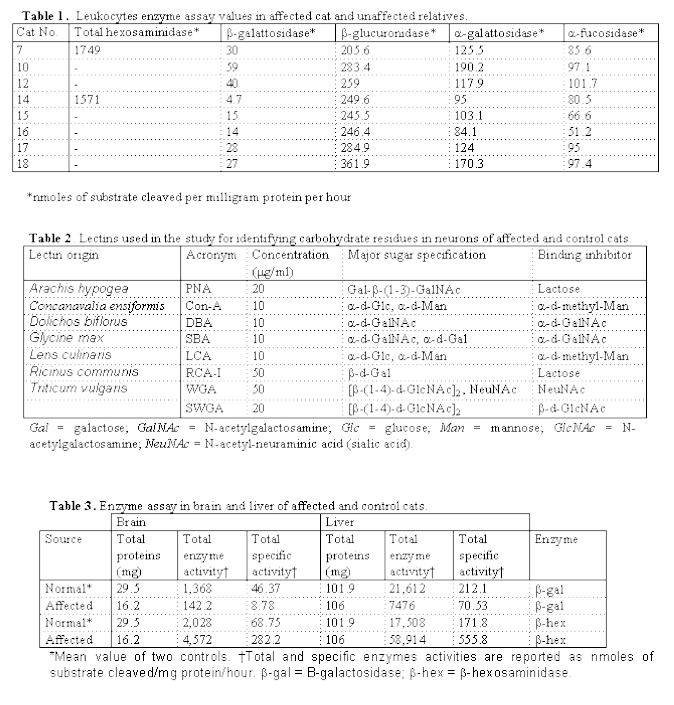
Abstract 7-month-old Korat cat was referred for a slowly-progressive neurological disease. Circulating monocytes and lymphocytes showed the presence of single or multiple empty vacuoles and blood leukocytes enzyme assay revealed a very low b-galactosidase activity level (4.7 nmol/mg/hr) as compared to unaffected parents and relatives. Histologically the cat, euthanized at the owner request at 21 months of age, presented diffuse vacuolization and enlargement of neurons throughout the brain, spinal cord and peripheral ganglia, severe cerebellar neuronal cell loss, and moderate astrocytosis. Stored material was stained with PAS on frozen sections and with RCA-I, Con-A and WGA lectins in paraffin wax-embedded sections. Ultrastructurally, neuronal vacuoles were filled with concentrically whorled lamellae and small membrane-bound vesicles. In the affected cat, b-galactosidase activity was markedly reduced in brain (18.9%) and l iver (33.25%) while total b-hexosaminidase activity showed a remarkable increase. Quantitation of total gangliosides revealed a 3-fold increase in brain and 1.7-fold in liver of affected cat. High-performance thin layer chromatography (HPTLC) detected a striking increase of GM1-ganglioside. On densitometric analysis of HPTLC bands, the absorption of GM1-ganglioside band was 98.52% of all stained bands (GD1a ,GD1b,GT1b ). Based on clinical onset, morphological and histochemical features, and biochemical findings, Korat cat GM1-gangliosidosis is comparable with the human type II (juvenile) form. However, clinical progression, survival time and level of b-galactosidase deficiency do not completely fit with those of human type II GM1-gangliosidosis. The disease in Korat cat is also different from other reported forms of feline GM1-gangliosidosis. Key words Gangliosidosis G(M1) € Feline lysosomal storage disease € Animal disease modelIntroduction Introduction GM1-gangliosidosis is an inherited lysosomal storage disease caused by deficient activity of lysosomal acid b-galactosidase resulting in accumulation of GM1-ganglioside and galactose-containing oligosaccharides in different cell types [25]. In humans, on the basis of age of clinical onset, evolution, and clinico-pathological findings, the disease is classified into three different forms, the infantile (type I), juvenile (type II) and chronic or adult (Type III) forms [21, 26, 37]. Type I form is characterized by early onset, rapid progression and severe neuroskeletal and visceral involvement. Type III form lacks of bony abnormalities and shows slowly progressive neurological symptoms and selective glycolipid accumulation in some brain areas [32, 40]. Type II form exhibits clinico-pathological features falling between type I and type III GM1-gangliosidosis. Although the clinico-pathological heterogeneity has been correlated with different b-galactosidase gene mutations [11, 14, 38], other genetic or environmental factors are thought to modify the phenotipic expression of GM1-gangliosidosis [33, 39] GM1-gangliosidosis has been identified in several animal species including cats [4, 7, 10], cattle [15], dogs [2, 27, 28], and sheep [30]. In sheep a b-galactosidase deficiency associated with a-neuraminidase deficiency also has been reported [1]. In Korat cats another form of gangliosidosis similar to Sandhoff¹s disease (GM2-gangliosidosis type II) of humans has also been identified [24].In this report, we describe the clinico-pathological and biochemical features of a new form of feline GM1-gangliosidosis in a Korat cat due to the deficient activity of b-galactosidase. Case report A seven-month-old, male, Korat cat was referred to the Veterinary Clinic of the Department of Animal Pathology, University of Turin, for retarded growth and a two weeks history of hind limb tremors and mild dyspnea. Anamnestic data revealed regurgitation episodes. Further anamnestic investigation showed that a kitten of a previous litter with neurological signs had died at the age of 7 months from severe gastro-enteritis. In the last three days, the owner reported the occurrence of seizures. Neurological examination revealed a depressed mental status, normal cranial nerve reflexes, ataxia with hypermetria, and spinal nerve hypereflexia. The postural reaction was normal except for the wheelbarrowing test, which showed slow initiation and hypermetria. Ophthalmoscopic examination showed no changes. Blood and biochemical profiles were within normal limits. One month later, the cat was re-examinated because of progressive paraparesis. Based on history and clinical signs a form of gangliosidosis was suspected. An enzymatic assay performed on blood leukocytes revealed a very low b-galactosidase activity level (4.7 nmol/mg/hr) in the diseased cat while total b-hexosaminidase, b-glucuronidase, a-galactosidase and a-fucosidase activities were within normal range, as compared to unaffected relatives (Table 1). The cat was euthanized at the owner request when he was 21 months old. A short pedigree of affected cat is shown in Fig.1. Material and methods Morphological, lectin histochemical and immunohistochemical studies White blood cells pellets from affected cat (n.14), non-affected relatives (cat n. 12, 15, 16, 17, and 18), obligate heterozygotes (cat n. 7 and 10) and two age-matched controls were examined on May-Grumwald-Giemsa stained smears by light microscopy and ultrastructurally. Following post-mortem examination, the brain was cut along the planum sagittalis into two symmetrical parts.One hemisphere and part of spinal cord, liver, and kidney were immediately frozen and stored at 80°C for biochemical studies. From the remaining hemisphere and from spinal cord, liver, kidney, skeletal muscle, myocardium, and lung, representative tissue portions were sampled and fixed in 10% neutral phosphate-buffered formalin for histology and electron microscopy. For histological examination, tissue samples from affected cat and from two unaffected age-matched controls were processed routinely in paraffin wax and stained with HE, periodic-acid-Schiff (PAS), Luxol-fast-blue (LFB) and Bodian methods. Frozen tissue sections were also stained by PAS, oil-red-O, and Sudan black. Four mm thick, paraffin-embedded sections from brain and spinal cord were also used for lectin histochemistry and anti-GFAP immunohistochemistry. For lectin histochemistry, tissue sections were incubated in a 2% H2O2 solution to block the endogenous peroxidase activity. To reduce background staining, the sections were covered with filtered mouse liver powder solution at a concentration of 150 mg/ml in phosphate-buffered saline (PBS), pH 7.2, for 15 min. Sections were then incubated for 30 min at room temperature with 8 different biotinylated lectins (Sigma-Aldrich Srl, Milano, Italy), washed twice in PBS and covered with avidin-biotin-peroxidase complex (ABC) (Vector Labs, Burlingame, California) for 30 min at room temperature. An equal amount of the lectin and 0.2M of the corresponding blocking sugar were incubated for 1 hour at room temperature before applying the solution to the sections; this incubation served as a control for binding specificity, and incubation of ABC alone served as a non-specific negative control. The lectin used in this study, their acronyms, the lectin concentrations used, their major sugar specificity, and the corresponding blocking sugars are listed in table 2. Anti-GFAP immunohistochemistry was performed by incubating paraffin-embedded sections with a policlonal antibody (6F2, DAKO S.p.A, Milano, Italy) 1:2000 for 45 min at 37°C after the block of endogenous peroxidase activity as above mentioned. Sections were covered with ABC complex (Vector Labs) for 1 hour at room temperature. For both lectin histochemistry and anti-GFAP immunohistochemistry sections were finally incubated with a solution of 0.01% diaminobenzidine and 3% H2O2 at room temperature for 4-8 min and lightly counterstained with Harris haematoxylin. Positively stained structures were demonstrated by the golden dark-brown 3.3-diaminobenzidine tetrahydrochloride-H202 reaction product. Brain and spinal cord paraffin-embedded sections from two age-matched European cats were used as a control for lectin histochemistry and anti-GFAP immunohistochemistry. For ultrastructural studies tissue samples from brain, spinal cord, peripheral ganglia, liver and kidney were post-fixed in 1% osmium tetraoxide in 0.1M cacodylate buffer at a pH of 7.4 and stained en bloc with 5% aqueous uranyl acetate. Blocks were dehydrated in graded ethanols solutions and embedded in Epon/Araldite (1:1). One mm thick sections were stained with toluidine blue and examined. Thin sections were cut 50 to 70 nm, stained by uranyl acetate and lead citrate and observed on a Zeiss EM109 with trans-fiber-optic photography. Leukocyte and serum enzyme analysis Blood samples were taken from the affected cat (n.14), unaffected relatives (cat n. 12, 15, 16, 17, and 18), obligate heterozygotes (cat n. 7 and 10) and two age-matched controls. Leukocytes were prepared from heparinized whole blood. Erythrocytes were sedimented with dextran (Mr 150,000 from Sigma-Aldrich), and the leukocytes-rich supernatant was centrifuged at 600 g for 10 min. The pellet obtained was resuspended in distilled water for 30-40 s and NaCl was added to the final concentration of 0.9% to lyse red blood cells. After centrifugation, the leukocyte-rich pellet was resuspended and sonicated before enzyme assay. The protein content of leukocyte samples was determined utilizing protein-dye binding (Bio-Rad Laboratories, srl, Milano, Italy) [12]. The substrate for leukocytes enzyme activity were 1.5mM of 4-methylumbrelliferyl-6-sulpho-2-acetamido-2-deoxy-b-d-glucopyranoside (Sigma-Aldrich) for b-hexosaminidase, 1mM of 4-MU-b-galactopyranoside (Sigma-Aldrich) for b-galactosidase, 5mM of 4-MU-b-d-glucuronide (Sigma-Aldrich) for b-glucuronidase, and 1mM of 4-MU-a-L-fucopyranoside (Sigma-Aldrich) for a-fucosidase. Substrate hydrolysis was measured spectrofluorometrically at an excitation wavelength of 360 nm and an emission wavelength of 450 nm. Specific activities were reported as nmoles of substrate cleaved per milligram protein per hour. Serum analysis for the presence of b-hexosaminidase isoenzymes was carried out with fluorescent staining of the bands with 2 mM 4-MU-2-acetamido-2-desossi-b-glucopyranoside after electrophoretic separation on cellulose acetate sheets in 0.025 M citric acid-sodium citrate, pH 5.5. Tissue samples enzyme assay Brain and liver samples from affected cat and two age-matched normal controls were homogenized in a 0.25 M sucrose, 5 mM Tris, and 1mM EDTA, solution at pH 7.5. Samples were then centrifuged at 10,000 g for 30 min at 4°C. Supernatant was removed, aliquoted and stored at 80°C. Crude homogenate was analyzed for total b-hexosaminidase and b-galactosidase activities with 1 mM 4-methylumbelliferyl-2-acetamido-2-deoxy-b-d-glucopyranoside and 1mM 4-methylumbelliferyl-b-d-galactopyranoside, respectively in 0.1 M acetate buffer, 0.125% Triton X-100, and 0.04% BSA, pH 4.4. Analysis was carried out using a modification of Storrie and Madden method [31] at pH 4.4. Substrate hydrolysis was analyzed spectrofluorometrically at an excitation wavelength of 364 nm and an emission wavelength of 448 nm. Specific activities were reported as nmoles of substrate cleaved per milligram protein per hour. Lipids and total glycosaminoglycans (GAG) analysis One gr of brain and liver samples from affected cat and two age-matched normal controls were homogenized (Polytron) in 4 ml of tetrahydrofuran (Sigma-Aldrich) and 1 ml of 10 mM phosphate buffer, pH 6.8. After centrifugation at 5000 g for 15 min at 4°C, supernatants was mixed with ether (3:1) and centrifuged again at 5000 g for 10 min at 4°C. Upper- and lower-phases (UP, LP) were removed and concentrated under vacuum (rotavapor). LP was dialyzed overnight against distilled water at 4°C, lyophilized and resuspended in chloroform/methanol (2:1) [36]. N-acetylneuraminic acid (NANA) content of LP was determined by the method of Svennerholm [34] and expressed as nmol/mg of wet tissue. High performance thin-layer chromatography (HPTLC) was performed on 20 x 20-cm silica gel HPTLC (Bracco Spa, Milano, Italy). Mobile phase was constituted by chloroform/methanol/0.2% calcium chloride (50:42:11) for LP and chloroform/methanol/ammonia (80:20:2) for UP . TLC of LP was developed by the resorcinol sulphuric acid reaction [34] while TLC of UP was stained with 0.3% ninhydrin in a butilic alcohol and 0.03% acetic acid solution. A densitometric analysis was also carried out on HPTLC stained bands (GS-700 Imaging Densitometer, Bio-Rad Laboratories). The relative absorption of the GM1-ganglioside band out of the total stained ganglioside bands was expressed as a percentage. For total glycosaminoglycans quantification, brain and liver samples were homogenized in 2 volumes of distilled water. Proteins were digested twice by incubation on each occasion with papain (1mg/1 ml of homogenized sample) (Boehringer Mannheim Italia S.p.A, Milano, Italy) for 48 hours at 40°C. A few drops of octanoic acid were added to prevent bacterial growth. After centrifugation at 12,000 g for 20 minutes, the supernatant was brought to 7% trichloroacetic acid, held for 1 hour at 4°C and centrifuged (12,000 g for 20 minutes). The supernatant was diluted with 4 volumes of cold ethanol (containing 1% potassium acetate), stored overnight at -20°C and centrifuged at 12,000 g for 10 min. Precipitated GAG were finally dissolved in distilled water proportional to the wet weight of MV (1ml/1gr). GAG were quantified by the carbazole method [9] using glucuronolactone as standard. The concentration was expressed as µg of uronic acid per gr of wet tissue. Results Morphological and lectin histochemical studies Light microscopic observation of leukocyte-rich pellet from the diseased cat, revealed the presence in circulating monocytes and in some lymphocytes of single or multiple empty vacuoles. Ultrastructurally membranebound vacuoles were mostly empty or filled with fine fibrillo-granular material or small vesicles (Fig. 2). Occasionally lamellated membrane structures were also observed. Vacuoles were not present in unaffected relatives. On post-mortem examination, no significant gross lesions, including organomegaly or skeletal abnormalities, were found. Histological observation revealed a diffuse vacuolization and enlargement of neurons throughout the brain, spinal cord, spinal and peripheral ganglia. All areas in the central nervous system (CNS) were severely affected except the granular layer of the cerebellum, ependymal and choroid plexus cells. Severe neuronal cell loss was detected in the cerebellar cortex (Purkinje and granular layer cells) and mid brain. Neuronal cell vacuolization was associated in many neuroanatomic areas with moderate astrocytosis, which was readily detected in sections immunostained for GFAP (Fig. 3). No inflammatory reaction was observed in any part of the CNS. Affected neurons showed a marked increase in size, a decrease in cytoplasmic staining intensity with a pale foamy or light-pink granular appearance, frequent nuclear margination, and clear marked cell borders. Stored material was also observed in axons. In some neurons, Nissl substance was present in a restricted perinuclear area where in others it was completely lost. In spinal cord some neurons showed single or multiple large empty vacuoles. On paraffin wax-embedded sections stained with PAS the content of vacuoles was stained only in some neurons located in medulla oblongata and in organs and in peripheral ganglia. In liver, almost all hepatocytes, regardless their position in the lobule showed extensive cytoplasmic vacuolization with a large single vacuole or multiple smaller vacuoles. Foamy vacuolization in kidney was mainly localized in the cortex with proximal and distal tubules, and glomerular podocytes diffusely involved while cortical collecting ducts and medullary tubular portions seemed to be unaffected. Vacuolization was observed also in epithelial cells of large bronchi, oesophagus, and adrenal medulla. In resin 1 mm thick sections, the content of neuronal vacuoles was observed as blue to dark blue stained granular material (Fig. 6). No such inclusions were seen in other cell types. Ultrastructurally, the cytoplasm of cerebral and spinal cord neurons, cerebellar Purkinje cells, and of peripheral ganglia were almost completely filled by membrane-bound irregularly shaped vacuoles ranging in diameter from 0.43 to 1.9 mm in diameter (Fig. 7). In these cells, cytoplasmic organelles were hardly observed. The stored material inside vacuoles was most commonly composed by irregularly parallel lamellated structures or concentrically whorled lamellae. However in largest vacuoles smaller membrane-bound vesicles with heterogeneous content were also frequently seen mixed with lamellated membrane structures (Fig. 7). The diameter of the smaller vesicles ranged from 0.26 to 0.5 mm. Stored fibrillo-granular material in hepatocytes was observed in membrane-bound vacuoles ranged 1.23 to 11.6 mm in size. Although in smaller vacuoles, the same type of stored material was also observed in Kupfer cells. Vacuolization in renal tubular cells involved all nephron tracts with particular intensity in proximal and distal tubules while in the renal glomerulus, podocytes, endothelial, and Bowman's capsule cells variable degree of cytoplasm vacuolization was found. In all kidney affected cells, the vacuoles content was of the fibrillo-granular type. Biochemical studies The results of leukocytes enzyme assay on affected and unaffected relatives are summarized in table 1. In the affected cat (n.14) a very low b-galactosidase activity (4.7) was found. The electrophoretic analysis of b-hexosaminidase isoenzymes revealed the presence of two bands corresponding to human hexosaminidase A and B, in all cats tested.< The results of enzyme assay in brain and liver of affected and control cats are summarized in table 3. Beta-galactosidase activity in the affected cat showed a marked decrease in brain (18.9%) and liver (33.25%) while total b-exosaminidase activity revealed a remarkable increased activity, compared to control cats. Identification of brain total gangliosides, as determined by the levels of lipid-bound N-acetyl-neuraminic acid (NANA), gave a mean value of 2,600 in normal cats and 7,790 in affected cat. In liver, total gangliosides were 100 in normal cats and 170 in affected cat. High-performance thin layer chromatography (HPTLC) analysis showed a striking increase of GM1-ganglioside in brain of affected Korat cat (Fig.8). Densitometric analysis of HPTLC GM1-ganglioside bands in normal and diseased cats was 54.55% and 98.52% respectively of the total absorption of all stained gangliosides bands (GD1a,GD1b,and GT1b). Total GAG quantification in brain and liver revealed a significant increase in the affected cat (147.7 and 3,816.3 respectively) compared to normal controls (29.5 and 731.42). Discussion The deficiency of acidic b-galactosidase and the accumulation of GM1-ganglioside in the brain and liver of the affected cat is consistent with the diagnosis of GM1-gangliosidosis. A preliminary analysis of the pedigree of affected cat indicates that the disease may have been inherited as a recessive autosomal trait since it originated with a consanguineous breeding and both parents are phenotypically normal. One sibling may have suffered of a neurological disease with clinical onset and features similar to those observed in the affected cat. Enzyme activity levels determined in circulating leukocytes from obligate heterozygotes and unaffected relatives seem not to help in identifying carriers. Direct identification of the mutation may provide a future tool to detect disease carrier. Although Baker et al. reported to know the occurrence of a case of GM1-gangliosidosis in a Korat cat [ 5], biochemical and clinico-pathological features of the disease in Korat cat were completely lacking. Based on clinical onset, morphological features and biochemical findings, this progressive neurological disorder seems to be comparable with human type II (juvenile) GM1-gangliosidosis. However, clinical progression, survival time, and level of b-galactosidase deficiency in Korat cat do not completely fit with those of human type II GM1-gangliosidosis [18, 23, 25, 33]. As concluded for the canine models of GM1-gangliosidosis [3], these findings indicate that it may not be completely appropriate to apply human classification to this animal model. Although a condition similar to human type II GM1-gangliosidosis with b-galactosidase deficiency and GM1-ganglioside accumulation has been described in Siamese [4] cats, significant clinico-pathological and biochemical differences are present with the disease in the Korat cat. Clinically, Siamese cats with GM1-gangliosidosis showed i mpaired vision, which was not evident in our case, and a more rapid progression of the disease (death by 8-12 months) compared to the affected Korat cat [4, 5, 6]. The longer duration of the disease may account for neuronal loss and astrocytosis found in brain of affected Korat cat which are not reported as characteristic in the Siamese cat model [17]. The longer duration of the disease may also explain the higher amount of stored GM1-ganglioside in brain associated with a higher b-galactosidase activity assayed in brain and liver of Korat cat as compared to Siamese cats [4, 17, 19]. All these findings indicate that GM1-gangliosidosis in Korat cat is different from that reported in other cat breed. HPTLC and GAG quantification detected a marked storage of GM1-ganglioside in brain and of GAG both in liver and brain indicating that enzymatic defect involve the catabolism of lipid- and protein-bound sugars. Data obtained from PAS reaction and lectin histochemistry on neuronal cells indicate the accumulation of glycolipids and oligosaccharides containing a-mannose-, b-galactose-, sialic acid-, and a-GalNAc-containing oligosaccharides [29]. All these sugar residues are normally present in N-linked oligosaccharides and in the GM1-ganglioside molecule [41]. Ultrastructural observations showed a clear difference in the stored material between neuronal cells and other cell types. In neurons enlarged lysosomes are mostly filled with lamellated structures, indicative of lipid accumulation, while in circulating leukocytes, hepatocytes, and kidney glomerular and tubular cells empty vacuoles or fibrillo-granular-filled vacuoles were detected , suggestive of stored oligosaccharides or glycosaminoglycans [16]. Although gangliosides are normal constituents of the plasma membrane, the amount and type of gangliosides in different tissue and in specific tissue cells is variable [41]. Hence, in liver and kidney epithelial cells, stored material is probably composed mainly of oligosaccharides rather than gangliosides which are more abundant in neuronal cells [35]. Neuronal cell loss was particularly severe in the cerebellum involving mainly Purkinje and granular layer cells. It has been shown that the cellular accumulation of sphingolipids has an inhibitory effect on protein kinase C with consequent progressive dysfunction of signal trasduction mechanisms leading to cell death [19]. Furthermore, a recent report has demonstrated that ceramide, which is part of the ganglioside molecule, is involved in the triggering of cardiomyocyte apoptosis [8]. Both series of data indicate that the accumulation of GM1-ganglioside may have initiated multiple biochemical events leading to the neuronal cell loss observed in affected Korat cat. Finally, lysosomal a-neuraminidase activity was not assayed because it is highly sensitive to freezing and thawing [22], and fresh tissue was not available in this case. Therefore a possible involvement of a-neuraminidase, reported in other lysosomal storage diseases [1], cannot be completely excluded. Acknowledgements The Authors are thankful to Ms. Patrizia Morra for her precious technical assistance. This work was supported by grants from M.U.R.S.T. (60%-1996). References 1. Ahern-Rindell AJ, Prieur DJ, Murname RD, Raghavan SS, Daniel PF, McCluer RH, Walkley SU, Parish SM (1988) Inherited lysosomal storage disease associated with deficiencies of b-galactosidase and a-neuraminidase in sheep. Am J Med Genet 31:39-56 2. Alroy J, Orgad U, Ucci AA, Schelling SH, Schunk KL, Warren CD, Raghavan SS, Kolodny EK (1985) Neurovisceral and skeletal GM1-gangliosidosis in dogs with beta-galactosidase deficiency. Science 229:470-472 3. Alroy J, Orgad U, DeGasperi R, Richard R, Warren CD, Knowles K, Thalhammer JG, Raghavan SS (1992) Canine GM1-gangliosidosis. A clinical, morphologic, histochemical, and biochemical comparison of two different models. Am J Pathol 140:675-689 4. Baker HJ, Lindsey JR, McKhann GM, Farrell DF (1971) Neuronal GM1 gangliosidosis in a Siamese cat with b-galactosidase deficiency. Science 174:838-839 5. Baker HJ, Mole JA, Lindsey JR, Creel RM (1976) Animal models of human ganglioside storage diseases. Fed Proc 35:1193-1201 6. Baker HJ, Reynolds GD, Walkley SU, Cox NR, Baker GH (1979) The gangliosidosis: comparative features and research applications. Vet Pathol 16:635-649 7. Barker CG, Blakemore WF, Dell A, Palmer AC, Tiller PR, Winchester BG (1986) GM1-gangliosidosis (type 1) in a cat. Biochem J 235:151-158 8. Bielawska AE, Shapiro JP, Jiang L, Melkonyan HS, Piot C, Wolfe CL, Tomei LD, Hannun YA, Umansky SR (1997) Ceramide is involved in triggering of cardiomyocyte apoptosis induced by ischemia and reperfusion. Am J Pathol 151:1257-1263 9. Bitter T, Muir HM (1962) A modified uronic acid carbazole reaction. Anal Biochem 4:330-334 10. Blakemore WF (1972) GM1-gangliosidosis in a cat. J Comp Path 82:82:179-185 11. Boustany R-M, Qian W-H, Suzuki K (1993) Mutations in acid b-galactosidase cause GM1-gangliosidosis in american patients. Am J Hum Genet 53:881-888 12. Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 7:248-254 13. Castagnaro M, Alroy J, Ucci AA, Glew RH (1987) Lectin histochemistry and ultrastructure of feline kidney from six different storage diseases. Virchows Arch B 54:16-26 14. Chakraborty S, Rafi MA, Wenger DA (1994) Mutations in the lysosomal b-galactosidase gene that cause the adult form of GMI gangliosidosis. Am J Hum Genet 54:1004-1013 15. Donnelly WJC, Sheahan BJ, Kelly M (1973) Beta-galactosidase deficiency in GM1- gangliosidosis of Fresian calves. Res Vet Sci 15:139-141 16. Dustin P, Tondeur M, Libert J (1978) Metabolic and storage diseases. In: Johannessen JV (ed) Electron microscopy in human medicine. McGraw-Hill Book Co, New York, pp 149-245 17. Farrell DF, Baker HJ, Herndon RM, Lindsey JR (1973) Feline GM1 gangliosidosis: biochemical and ultrastructural camparisons with the disease in man. J Neuropath Exp Neurol 32: 1-18 18. Giugliani R, Coelho Dutra J, Saraiva Pereira ML, Rotta N, de Lourdes Drachler M, Ohlweiller L, Monteiro de Pina Neto J, Pinheiro CE, Breda DJ (1985) GM1 gangliosidosis: clinical and laboratory findings in eight families. Hum Genet 70:347-354 19. Handa S, Yamakawa T (1971) Biochemical studies in cat and human gangliosidosis. J Neurochem 18:1275-1280 20. Hannun YA, Bell RM (1987) Lysosphingolipids inhibit protein kinase C: implications for the sphingolipidosis. Science 235:670-674 21. Kasama T, Taketomi T (1986) Abnormalities of cerebral lipids in GM1-gangliosidosis, infantile, juvenile, and chronic type. Jpn J Exp Med 56:1-11 22. Knowles K, Alroy J, Castagnaro M, Raghavan SS, Jakowski RM, Freden GO (1993) Adult-onset lysosomal storage disease in a Schipperke dog: clinical, morphological and biochemical studies. Acta Neuropathol 86:306-312 23. Mutoh T, Naoi M, Takahashi A, Hoshino M, Nagai Y, Nagatsu T (1986) Atypical adult GM1-gangliosidosis: biochemical comparison with other forms of primary b-galactosidase deficiency. Neurol 36:1237-1241 24. Neuwelt EA, Johnson WG, Blank NK, Pagel MA, Maslen-McClure C, McClure MJ, Wu PM (1985) Characterization of a new model of GM2-gangliosidosis (Sandhoff's disease) in Korat cats. J Clin Invest 76:482-490 25. O'Brien JS (1983) The gangliosidoses. In: Stanbury JB, Wyngaarden JB, Fredrickson DAS, Goldstein JL, Brown MS (eds) The metabolic bases of inherited diseases, 5th edn. McGraw-Hill Book Co, New York, pp 592-599 26. O'Brien JS (1989) b-Galactosidase deficiency (GM1 gangliosidosis, galactosialidosis, and Morquio syndrome type B); ganglioside sialidase deficiency (mucolipidosis IV). In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic bases of inherited diseases, 6th edn. McGraw-Hill Book Co, New York, pp 1797-1806 27. Read DH, Harrington DD, Keenan TW, Hinoman EJ (1976) Neuronal-visceral GM1-gangliosidosis in a dog with b-galactosidase deficiency. Science 194:442-445 28. Saunders GK, Wood PA, Myers RK, Shell LG, Carithers R (1988) GM1 gangliosidosis in Portuguese water dogs: pathologic and biochemical findings. Vet Pathol 25:265-269 29. Sharon N, Lis H (1989) Carbohydrate specificity. In: Sharon N, Lis H (Eds) Lectins. Chapman and Hall, London, pp 36-46 30. Skelly BJ, Jeffrey M, Franklin RJM, Winchester BG (1995) A new form of ovine GM1-gangliosidosis. Acta Neuropathol (Berl) 89:374-379 31. Storrie B, Madden EA (1990) Isolation of subcellular organelles. Methods Enzymol 182:203-220 32. Suzuki K (1991) Neuropathology of late onset gangliosidosis. A review. Dev Neurosci 13:205-210 33. Suzuki Y, Sakuraba H, Oshima A, Yoshida K, Shimmoto M, Takano T, Fukuhara Y (1991) Clinical and molecular heterogeneity in hereditary b-galactosidase deficiency. Dev Neurosci 13:299-303 34. Svennerholm L (1957) Quantitative estimation of sialic acids. II. A colorimetric resorcinol-hydrochloric acid method. Biochim Biophys Acta 24:604-611 35. Sweeley CC, Siddiqui B (1977) Chemistry of mammalian glycolipids. In: Horowitz MI, Pigman W (eds) The glicoconjugates, vol 1. Academic Press, New York, pp 459-540 36. Tettamenti G, Bonali F, Marchesini S, Zambotti V (1973) A new procedure for extraction, purification and fractionation of brain gangliosides. Biochim Biophys Acta 296:160-170 37. Warner TG, Robertson AD, O'Brien JS (1983) Diagnosis of GM1 gangliosidosis based on detection of urinary oligosaccharides with high performance liquid chromatography. Clin Chim Acta 127:313-326 38. Yoshida K, Oshima A, Shimmota M, Fukuhara Y, Sakuraba H, Yanagisawa N, Suzuki Y (1991) Human b-galactosidase gene mutations in GM1-gangliosidosis: a common mutation among Japanese adult/chronic cases. Am J Hum Genet 49:435-442 39. Yoshida K, Oshima A, Sakuraba H, Nakano T, Yanagisawa N, Inui K, Okada S, Uyama E, Namba R, Kondo K, Iwasaki S, Takamiya K, Suzuki Y (1992) GM1 gangliosidosis in adults: clinical and molecular analysis of 16 Japanese patients. Ann Neurol 31:328-332 40. Yoshida K, Ikeda S, Kawaguchi K, Yanagisawa N (1994) Adult GM1 gangliosidosis: immunohistochemical and ultrastructural findings in an autopsy case. Neurol 44:2376-2382 41. Yu RK, Saito M (1989) Structure and localization of gangliosides. In: Margolis U, Margolis RK (eds) Neurobiology of glycoconjugates. Plenum Press, New York, pp 1-42 |
||
 |
||
Legend for figures Table 1 Fig.1 - Pedegree of Korat cat with GM1-gangliosidosis. Cat n. 11 had suffered of an undiagnosed progressive neurological disease. Fig.2 - Electron micrograph showing cytoplasmic membrane-bound vacuoles with fibrillo-granular material in a circulating monocyte. Bar = 0.8 mm. Table 2 Fig.3 - Cerebral cortex showing moderate astrocytosis. Anti-GFAP and hematoxylin stain. Bar = 25 mm. Fig.4 - Spinal chord. Anterior horn neurons show strong Periodic acid-Schiff (PAS) reaction in their cytoplasms. PAS stain. Bar = 200 mm. Fig.5 - Spinal chord. Moderate to strong cytoplasmic reaction for RCA-I lectin is evident in the cytoplasm of many neurons. RCA-I stain. Bar = 100 mm. Fig.6 - Cerebral cortex. Both neurons and an enlarged axon show granular intracytoplasmic inclusions of various staining intensity. Resin-embedded one mm thick section, toluidine blue stain. Bar = 10 mm. Table 3 Fig. 7 - Electron micrograph showing cytoplasmic membrane-bound vacuoles with whorled lamellated and vesicular content. Bar = 0.3 mm. Fig. 8 - Silica gel high performance thin layer chromatography (HPTLC) of brain gangliosides of controls (lane 1and 2) and affected Korat cat (lane 3 and 4), and standard GM1-, GM2-, and GM3-gangliosides (GM1, GM2, GM3).
© Massimo Castagnaro © |
||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||


Beta-galactosidase deficiency in a Korat cat:
a new form of feline GM1-gangliosidosis.